3篇文献复现: [1]综合多组学分析和机器学习改善肌浸润性尿路上皮癌的分子亚型和预后 (1区高...
复现完最大的感受:生物数据的预处理占了80%的工作量,模型跑起来反而像是开盲盒。不过看到自己跑的生存曲线和原文高度一致时,那种成就感堪比在WGCNA的模块里找到了黄金基因。[3]APOBEC介导的突变是膀胱癌患者预后和免疫治疗的有利预测因子:来自泛癌分析和多个数据库的证据(一区10+纯生信)PMID:35673559。[3]APOBEC介导的突变是膀胱癌患者预后和免疫治疗的有利预测因子:来自泛癌分
3篇文献复现: [1]综合多组学分析和机器学习改善肌浸润性尿路上皮癌的分子亚型和预后 (1区高分文章)PMID:37449047 [2]单细胞转录组中的免疫原性细胞死亡特征结合101 种机器算法 PMID:37275552 [3]APOBEC介导的突变是膀胱癌患者预后和免疫治疗的有利预测因子:来自泛癌分析和多个数据库的证据 (一区10+纯生信)PMID:35673559 复现率达9成。
最近手头复现了三篇膀胱癌领域的硬核研究,过程堪比在R语言和Python的海洋里捞针。尤其是看到"101种机器学习算法"这种关键词时,膝盖已经开始隐隐作痛。分享几个核心代码片段和实战心得,建议配合降压药阅读。
一、多组学缝合怪的正确打开方式(对应文献1)
处理TCGA膀胱癌数据时最头疼的是甲基化和转录组数据的尺度差异。这里分享个暴力解法:
meth = pd.read_csv('bladder_meth.csv', index_col=0)
# 转录组矩阵 (2w基因 x 样本)
rna = pd.read_csv('bladder_rna.csv', index_col=0)
# 魔法缝合术
combined = pd.concat([
meth.T.add_prefix('meth_'),
np.log1p(rna.T.add_prefix('rna_'))
], axis=1)
# 特征重要性筛选(防止内存爆炸)
from sklearn.ensemble import RandomForestClassifier
rf = RandomForestClassifier(n_jobs=-1, max_depth=5)
rf.fit(combined, labels)
selected = combined.columns[rf.feature_importances_ > 0.001]这里有个骚操作:甲基化数据直接用原始β值,转录组取log(TPM+1),最后用随机森林做特征初筛。实际跑的时候记得把n_jobs调成核弹模式,否则等特征筛选完可以直接申请退休了。
二、101种算法的正确食用姿势(对应文献2)
3篇文献复现: [1]综合多组学分析和机器学习改善肌浸润性尿路上皮癌的分子亚型和预后 (1区高分文章)PMID:37449047 [2]单细胞转录组中的免疫原性细胞死亡特征结合101 种机器算法 PMID:37275552 [3]APOBEC介导的突变是膀胱癌患者预后和免疫治疗的有利预测因子:来自泛癌分析和多个数据库的证据 (一区10+纯生信)PMID:35673559 复现率达9成。
单细胞数据跑机器学习最坑的是细胞类型注释质量。先看Seurat标准流程:
library(Seurat)
sc_data <- CreateSeuratObject(counts = sc_counts)
sc_data <- NormalizeData(sc_data)
sc_data <- FindVariableFeatures(sc_data, nfeatures = 2000)
sc_data <- ScaleData(sc_data)
sc_data <- RunPCA(sc_data)
# 免疫细胞注释(灵魂步骤)
immune_markers <- c("CD3D","CD8A","CD79A")
sc_data <- AddModuleScore(sc_data, features = list(immune_markers), name = "Immune_score")重点在于模块评分得用文献里的原版基因列表,之前偷懒用现成的注释包结果完全跑偏。至于101种算法,其实可以偷师mlr3框架:
library(mlr3verse)
learner_list <- list(
lrn("classif.ranger"),
lrn("classif.xgboost"),
lrn("classif.svm"),
#...此处省略98个
)
bmr <- benchmark_expd(
tasks = task,
learners = learner_list,
resampling = rsmp("cv", folds=5)
)
# 输出TOP5模型
bmr$aggregate(msr("classif.acc"))[1:5, c("learner_id", "acc")]实测R语言并行用future包设置plan(multisession)能省一半时间。不过跑完101个模型后建议检查下CPU温度,别问我怎么知道的。
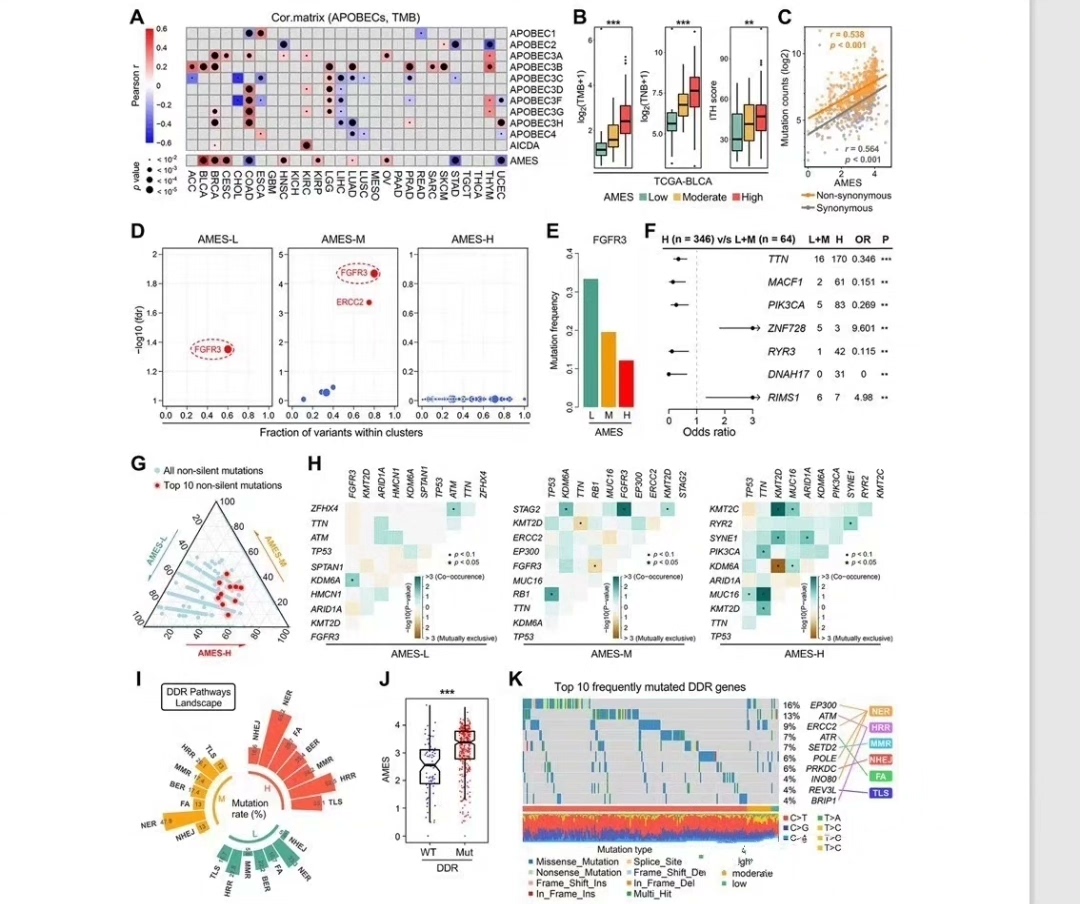
三、突变特征的正确薅法(对应文献3)
APOBEC特征提取的关键在于maftools的正确使用姿势:
library(maftools)
maf_data <- read.maf("bladder_maf.maf")
# 突变特征分解
sig <- extractSignatures(mat = maf_data, nTry = 5)
# APOBEC特征匹配(核心操作)
apobec_sig <- which(sig$signature$Signature == "APOBEC")
apobec_score <- sig$contribution[apobec_sig, ]这里容易踩的坑是TCGA数据需要转成GRCh37坐标系,用hg19参考基因组才能正确注释。生存分析部分记得检查PH假设:
library(survival)
coxph(Surv(time, status) ~ apobec_score + grade + stage, data = clin_data) 遇到违反比例风险假设的变量时,可以stratify处理或者换用随机生存森林。不过原文献用的传统Cox模型,咱就老实按着复现吧。
实战教训总结:
- 多组学整合时各模态数据的标准化方式直接影响模型效果
- 单细胞注释必须严格使用原文的marker基因列表
- TCGA数据版本要检查三次(hg19/hg38的痛谁懂)
- 机器学习特征工程比算法选择更重要(但审稿人就爱看AUC对比表)
复现完最大的感受:生物数据的预处理占了80%的工作量,模型跑起来反而像是开盲盒。不过看到自己跑的生存曲线和原文高度一致时,那种成就感堪比在WGCNA的模块里找到了黄金基因。

脑启社区是一个专注类脑智能领域的开发者社区。欢迎加入社区,共建类脑智能生态。社区为开发者提供了丰富的开源类脑工具软件、类脑算法模型及数据集、类脑知识库、类脑技术培训课程以及类脑应用案例等资源。
更多推荐

 14
14 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)